The “Big Picture”: For an overview of how our work on chromosomal stability connects to cellular disease states, check out our Research Overview page.
The big question: In 1902 Theodor Boveri posited that changes in chromosomes are linked to cancer. Decades of histology and molecular analysis of chromosomes have confirmed this linkage, though the causality of this linkage remains uncertain. At the heart of the issue is the question of how cancer cells make mistakes in segregating their chromosomes and do these errors contribute to either the onset or progression of cancer. We view this process of part of microevolutionary process that allows cells to adapt to a “disease state”.
Early work from my lab focused on understanding how cancer cells are altered to make errors in chromosome segregation that lead to aneuploidy (or hyperploidy) and how cells “evolve” the ability to tolerate such dramatic changes in their ploidy.
Errors in chromosome segregation are “early” in colorectal cancer models: Our initial work focused on understanding the causes of persistent errors in chromosome segregation. We focused on colorectal cancer as it represents a clear example of tumor cells that have a high error rate.
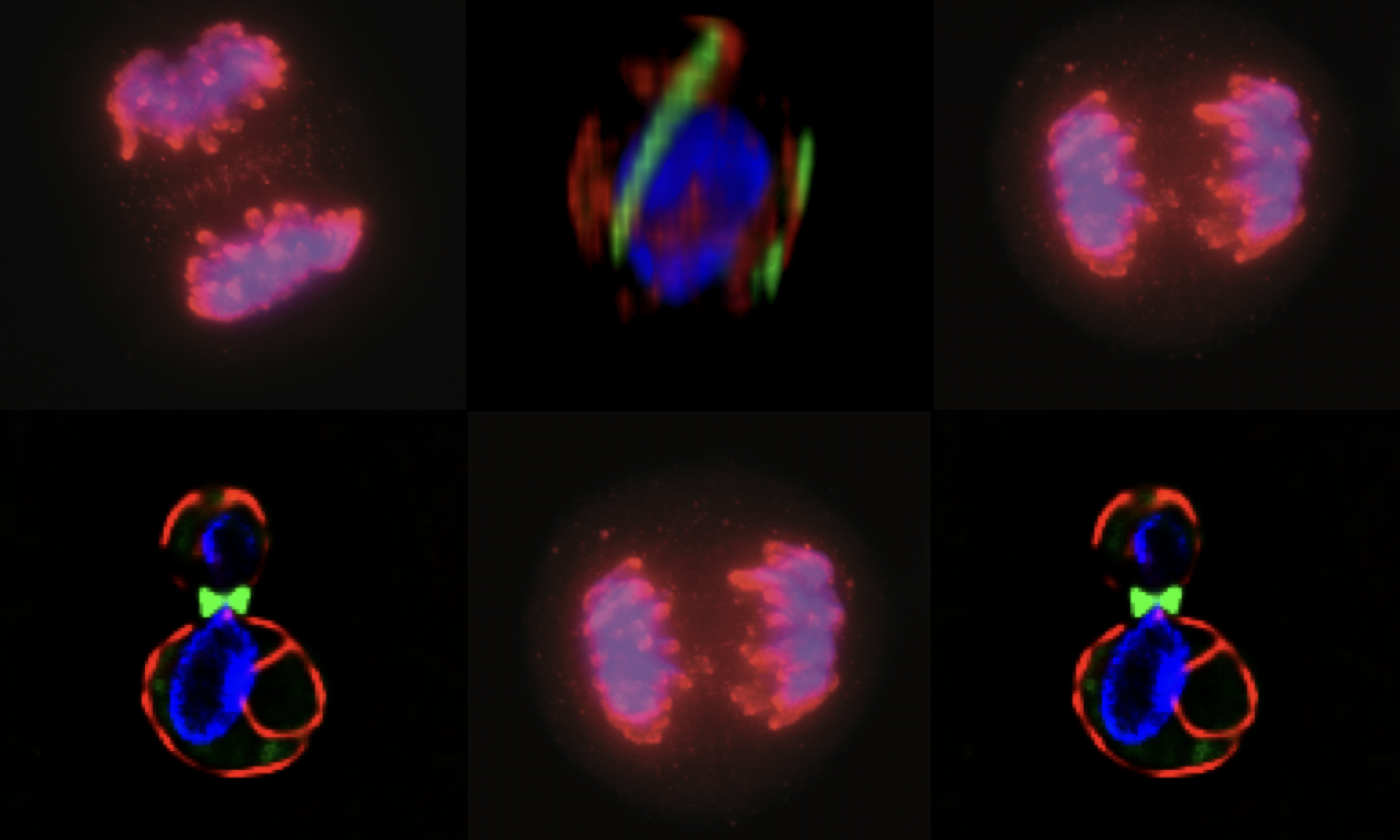
Adenomatous polyposis coli and mitotic errors: Adenomatous polyposis coli (APC) loss of heterozygosity is associated with colorectal cancer onset and progression. The recessive phenotype arising from loss of APC is the upregulation of the beta-catenin protein, associated with transcriptional reprogramming of the guts epithelium that favors cell division. In contrast to this recessive phenotype, we showed that a single mutant allele of APC is sufficient to dominantly induce mitotic errors. This implies that a single mutation in a tumor suppressor can dominantly drive cancer.
As shown in the figure below, canonical APC mutations found in colorectal cancer result in truncated proteins that can physically associated with the full length APC. This forms a toxic, or dominant negative, dimer (see cartoon) that specifically inhibits the ability of APC to regulate microtubule dynamics. The most dramatic dominant phenotype are mitotic spindle defects that lead to failures in chromosome attachment, spindle collapse and errors in chromosome segregation.
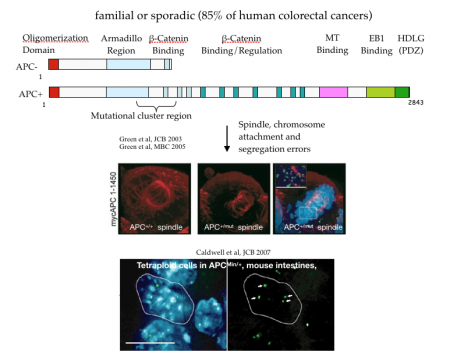
Do mitotic errors contribute to cancer onset or progression or are they a consequence of cancer adaptations? We do not know the answer to this important question. Our observations in mouse models suggest that the dominant activity of APC alleles can in principle cause mitotic errors in normal (or non-cancerous) cells. However, even in patients who inherit a single mutant APC from mom or dad, they do not exhibit cancer until their teens or early 20s, though they are “dominantly susceptible” to cancer. This suggests that any contribution of mitotic errors to cancer onset is slow and perhaps indirect. Moreover, a variety of studies from a diverse array of cells show that even a single extra chromosome, much less many extra chromosomes, cause a stress response that inhibits cell division. This paradox argues against a direct causal role for mitotic errors in cancer onset, though it doesn’t rule out the possibility that cells co-evolve “programs” that support aneuploidy and cell division, a state of instability that may further contribute to cancer onset or progression.
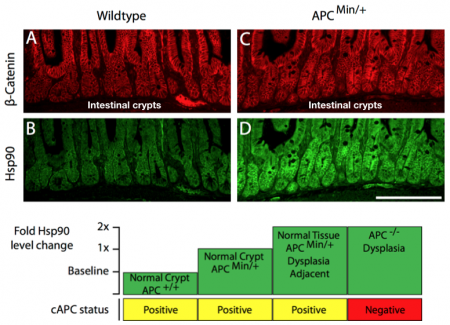
One way to rationalize the paradox of hyperploidy and cell fitness is to propose that cells are reprogrammed to “tolerate” extra chromosomes and associated proteostatic and genomic stressors that arise. Evidence to support such an “adaptive” reprogramming came from our observations of changes that occur in the intestinal crypts (the site where cancer initiates) of APC mutant mice that preceded the canonical markers of cancer onset. We observed that a single mutation in a tumor suppressor gene unregulated the proteostasis pathway under control of the transcription factor, Hsf1. We found that cytoskeletal stress caused by mutant APC could be sufficient to activate Hsf1 gene transcription. Moreover, we found increased levels of the Hsf1 target, Hsp90, in normal intestinal crypts harboring a single mutant APC allele. The levels of Hsp90 were additionally elevated in normal cells adjacent to early dysplastic regions and further elevated in the early cancer cells themselves (see figure below).
Future directions and the cancer “adaptive” state: Our findings raise the possibility that chronic cellular stressors – in the studies above induced by mutant APC – can lead to reprogramming of proteostatic pathways that allow cancer states to evolve. We further propose that multiple stress pathways re reprogrammed, including those that respond to genomic stress, allow cells to adapt to the burden associated with aneuploidy. We are now pursuing these ideas using a model system to mimic the cell stress associated with disease states. We predict that chronic stress can lead to reprogramming that (i) allows cancer cells to survive and proliferate or (ii) loss of cell homeostasis associated with neurodegenerative disease.